





Lisaks tehnoloogiale on glükosiidide süntees alati pakkunud huvi ka teadusele, kuna see on looduses väga levinud reaktsioon. Schmidti ja Toshima ning Tatsuta hiljutised artiklid ja paljud neis viidatud viited on kommenteerinud laia valikut sünteetilisi võimalusi.
Glükosiidide sünteesil kombineeritakse mitme suhkru komponente nukleofiilidega, näiteks alkoholide, süsivesikute või valkudega. Kui on vaja selektiivset reaktsiooni ühe süsivesiku hüdroksüülrühmaga, tuleb esimeses etapis kaitsta kõiki teisi funktsioone. Põhimõtteliselt võivad ensümaatilised või mikroobsed protsessid oma selektiivsuse tõttu asendada keerulisi keemilisi kaitsmise ja kaitserühmade eemaldamise etappe, et glükosiididest piirkondades selektiivselt eemaldada. Alküülglükosiidide pika ajaloo tõttu pole ensüümide kasutamist glükosiidide sünteesis aga laialdaselt uuritud ja rakendatud.
Sobivate ensüümsüsteemide mahutavuse ja kõrgete tootmiskulude tõttu ei ole alküülpolüglükosiidide ensümaatiline süntees tööstuslikule tasemele viidud ning eelistatud on keemilised meetodid.
1870. aastal teatas MAcolley "atsetoklorhüdroosi" (1, joonis 2) sünteesist dekstroosi (glükoosi) ja atsetüülkloriidi reageerimisel, mis viis lõpuks glükosiidide sünteesimise ajaloo alguseni.
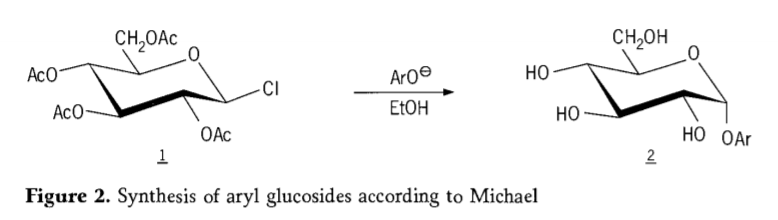
Tetra-O-atsetüülglükopüranosüülhalogeniidid (atsetohalogenoglükoosid) osutusid hiljem kasulikeks vaheühenditeks puhaste alküülglükosiidide stereoselektiivsel sünteesil. 1879. aastal õnnestus Arthur Michaelil valmistada Colley vaheühenditest ja fenolaatidest kindlaid, kristalliseeruvaid arüülglükosiide. (Aro-, joonis 2).
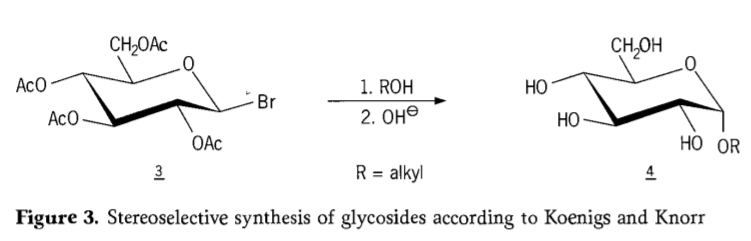
1901. aastal toimus Michaeli süntees laia süsivesikute ja hüdroksüülrühmadega aglükonide saamiseks, kui W. Koenigs ja E. Knorr tutvustasid oma täiustatud stereoselektiivset glükosideerimisprotsessi (joonis 3). Reaktsioon hõlmab SN2 asendust anomeersel süsinikul ja kulgeb stereoselektiivselt konfiguratsiooni inversiooniga, tootes näiteks α-glükosiidi 4 atseobromoglükoosi vaheühendi 3 β-anomeerist. Koenigsi-Knorri süntees toimub hõbeda või elavhõbeda promootorite juuresolekul.
1893. aastal pakkus Emil Fischer välja põhimõtteliselt teistsuguse lähenemisviisi alküülglükosiidide sünteesiks. See protsess on nüüd tuntud kui „Fischeri glükosideerimine“ ja see hõlmab glükooside happekatalüüsitud reaktsiooni alkoholidega. Igas ajaloolises ülevaates peaks siiski olema ka A. Gautier' esimene teadaolev katse 1874. aastal muuta dekstroos veevaba etanooliga vesinikkloriidhappe juuresolekul. Eksitava elementanalüüsi tõttu arvas Gautier, et ta oli saanud „diglükoosi“. Hiljem tõestas Fischer, et Gautier' „diglükoos“ oli tegelikult peamiselt etüülglükosiid (joonis 4).
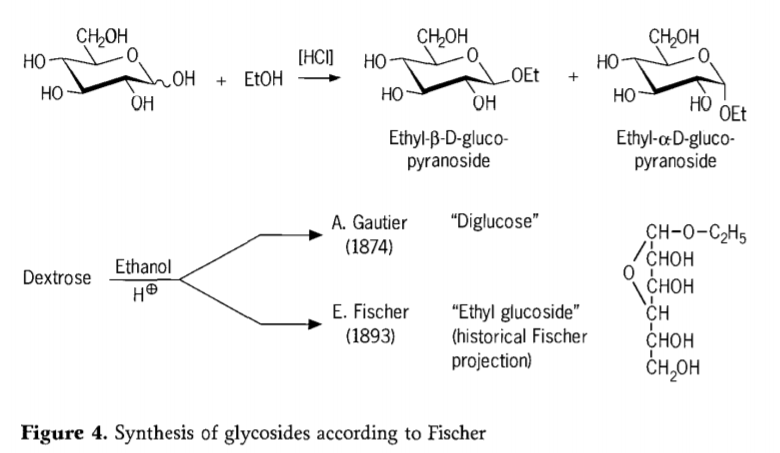
Fischer defineeris etüülglükosiidi struktuuri õigesti, nagu nähtub ajaloolisest furanosiidvalemist. Tegelikult on Fischeri glükosideerimisproduktid keerulised, enamasti α/β-anomeeride ja püranosiid/furanosiid isomeeride tasakaalulised segud, mis sisaldavad ka juhuslikult seotud glükosiidoligomeere.
Seega ei ole üksikuid molekulaarseid liike Fischeri reaktsioonisegudest lihtne eraldada, mis on varem olnud tõsine probleem. Pärast selle sünteesimeetodi mõningast täiustamist võttis Fischer oma uuringutes kasutusele Koenigs-Knorri sünteesi. Seda protsessi kasutades olid E. Fischer ja B. Helferich esimesed, kes teatasid 1911. aastal pindaktiivsete omadustega pika ahelaga alküülglükosiidi sünteesist.
Juba 1893. aastal märkas Fischer õigesti alküülglükosiidide olulisi omadusi, näiteks nende suurt stabiilsust oksüdatsiooni ja hüdrolüüsi suhtes, eriti tugevalt aluselises keskkonnas. Mõlemad omadused on alküülpolüglükosiidide jaoks väärtuslikud pindaktiivsete ainete rakendustes.
Glükosüdeerimisreaktsiooniga seotud uuringud on endiselt pooleli ja viimasel ajal on välja töötatud mitu huvitavat glükosiidide sünteesimise viisi. Mõned glükosiidide sünteesi protseduurid on kokku võetud joonisel 5.
Üldiselt võib keemilise glükosideerimise protsesse jagada protsessideks, mis viivad happe katalüüsitud glükosüülivahetuses keeruka oligomeeride tasakaaluni.
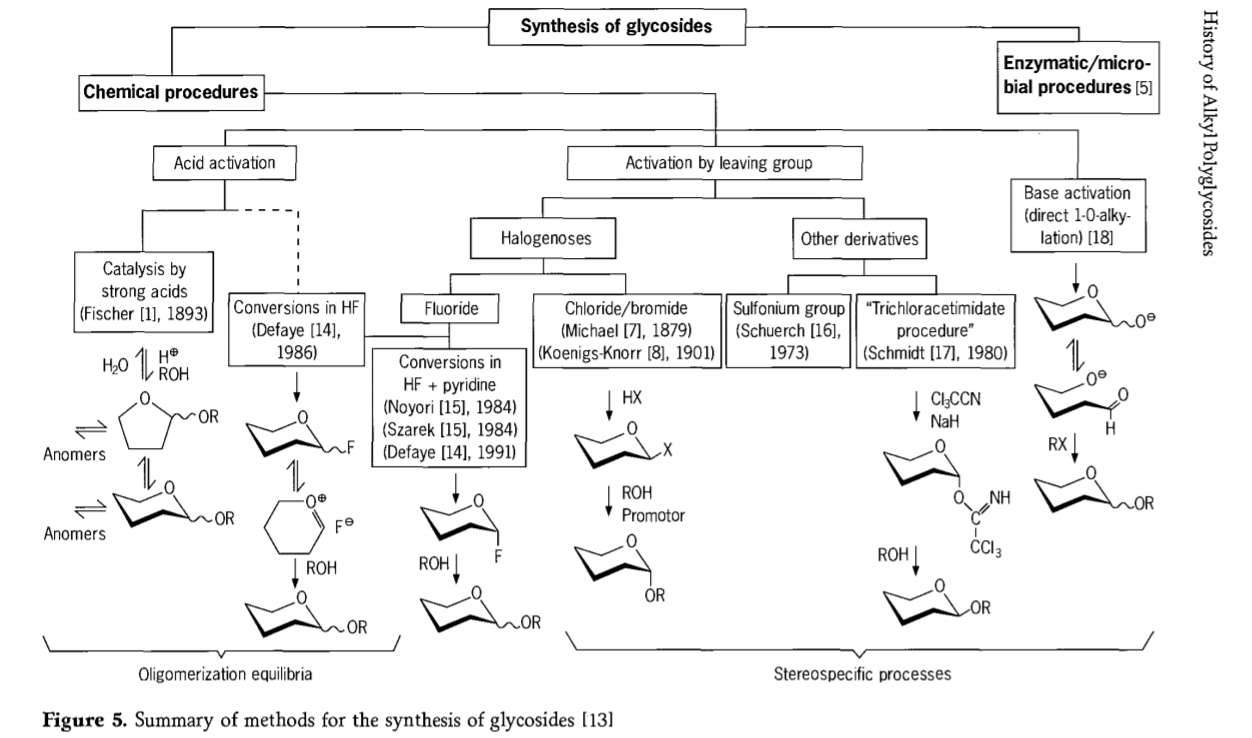
Reaktsioonid sobivalt aktiveeritud süsivesikute substraatidel (Fischeri glükosiidreaktsioonid ja vesinikfluoriidi (HF) reaktsioonid kaitsmata süsivesikute molekulidega) ja kineetikaga kontrollitud, pöördumatud ja peamiselt stereotaksilised asendusreaktsioonid. Teist tüüpi protseduur võib viia üksikute liikide moodustumiseni, mitte reaktsioonide keeruliste segude moodustumiseni, eriti kombineerituna konservatsioonirühmade tehnikatega. Süsivesikud võivad jätta rühmi ektoopilisele süsinikule, näiteks halogeeni aatomid, sulfonüülid või trikloroatseetimidaatrühmad, või olla aktiveeritud alustega enne triflaatestriteks muundamist.
Vesinikfluoriidis või vesinikfluoriidi ja püridiini segudes (püridiiniumpolü[vesinikfluoriid]) toimuvate glükosideerimiste puhul moodustuvad glükosüülfluoriidid in situ ja muunduvad sujuvalt glükosiidideks, näiteks alkoholidega. Vesinikfluoriid osutus tugevalt aktiveerivaks, mittelagunevaks reaktsioonikeskkonnaks; täheldati tasakaalulist autokondensatsiooni (oligomerisatsiooni), mis sarnanes Fischeri protsessiga, kuigi reaktsioonimehhanism on tõenäoliselt erinev.
Keemiliselt puhtad alküülglükosiidid sobivad ainult väga erilisteks rakendusteks. Näiteks on alküülglükosiide edukalt kasutatud biokeemilistes uuringutes membraanvalkude kristalliseerimiseks, näiteks poriini ja bakteriorodopsiini kolmemõõtmeliseks kristalliseerimiseks oktüül-β-D-glükopüranosiidi juuresolekul (sellel tööl põhinevad edasised katsed viisid Deisenhoferi, Huberi ja Micheli Nobeli keemiaauhinnani 1988. aastal).
Alküülpolüglükosiidide väljatöötamise käigus on laboris kasutatud stereoselektiivseid meetodeid mitmesuguste mudelainete sünteesimiseks ja nende füüsikalis-keemiliste omaduste uurimiseks. Nende keerukuse, vaheühendite ebastabiilsuse ning protsessijäätmete hulga ja kriitilise iseloomu tõttu tekitaksid Koenigs-Knorri tüüpi ja muud kaitserühma tehnikad olulisi tehnilisi ja majanduslikke probleeme. Fischeri tüüpi protsessid on suhteliselt vähem keerukad ja kaubanduslikus mahus lihtsamad läbi viia ning seetõttu on need eelistatud meetod alküülpolüglükosiidide tootmiseks suuremahulises mahus.
Postituse aeg: 12. september 2020